|
|
|
|
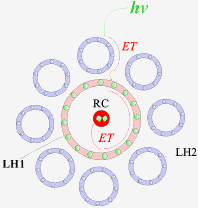
2. Light-harvesting complexes (LHC)Nature uses a variety of chromophores to harness solar energy towards chemical energy production. Several types of light-harvesting (LH) systems exist. For example, in purple bacteria, the LH1 complex, which contains protein-embedded chlorophyll and carotenoids, encircles the reaction centre (RC) (Fig. 3).4 Further removed are the LH2 complexes that feed excited-state energy into LH1. In some bacteria, a third LH3 complex exists. Energy is cascaded from LH3 and LH2 through LH1 to the RC with near unit efficiency. Duplicating this energy gradient and antenna-like disposition of chromophores is one of our synthetic targets and is highlighted in this section.
Figure 3 Schematic representation of light-harvesting complexes (LH1 and LH2) around the reaction center (RC). Arrows indicate the path of energy transfer (ET).4 2.1 Polymetallic DendrimersComplexes of Ru and Os are well-suited to act as chromophores in polymetallic dendrimers.5 In particular, the choice of using Ru and Os based chromophores as opposed to purely organic ones has been guided by the following:
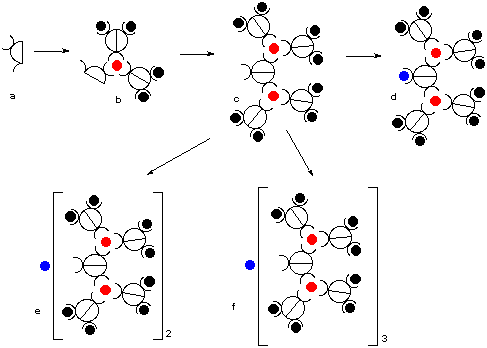
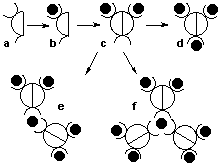
The photophysical properties of these dendrimers are examined in collaborations with groups in Europe (see Section 6). New collaborations concerning different aspects of this research will be initiated. 2.1.1 Dendrimers with metal ions at the branching pointsThe key methodology that we have developed is the creation of new metal-ion binding sites in metal complexes. As depicted in Figure 4, a ligand (Fig. 4, a) containing a bidentate binding site and an ortho-chloroimine site forms a ruthenium complex by selectively complexing a metal ion with octahedral coordination geometry in the free bidentate site of the ligand (Fig. 4, b). The ortho-chloroimine site in b is then coupled to create a new bidentate binding site (Fig. 4, c), which may in turn bind to another metal ion (Fig. 4, d), form a "complex-metal" (Fig. 4, e; c.f., Ru(bpy)2Cl2), or give a closed dendrimer (Fig. 4, f). Thus, in the first dendrimer generation, we have access to penta- and hepta-metallic complexes. The total metal ion content will grow more rapidly than with current approaches.5 Although many stereoisomers may exist, the differences in their electrochemical and spectroscopic properties are expected to be minimal.
Figure 4 Schematic representation of the synthesis of the first generation of polymetallic complexes Our approach requires fewer steps than current ones, such as the complexes-as-metals approach,6 which calls for an iterative protection/deprotection sequence. This latter approach consumes both time and materials. Another drawback to most systems is the slow increase in metal ion content, which requires several more steps to obtain dendrimers with high (>20) metal ion content. Indeed, very few well-characterized dendrimers exist with >20 metal ions.7
We have demonstrated the feasibility of our methodology using 4-chloro-(2-pyridyl)pyrimidine 1 as a ligand (c.f., Fig. 4a).6 It contains a bidentate pyridylpyrimidine site and a chloropyrimidine site. Metal complexation is favoured at the bidentate site. Thus, 4-chloro-(2-pyridyl)pyrimidine 1 was allowed to react with Ru(bpy)2Cl2.2H2O affording the red monometallic complex 2. It is important to note that no protection/deprotection sequence is required; the bidentate pyridylpyrimidine moiety is a better binding site than the monodentate, sterically encumbered ortho-halopyrimidine site. The monometallic complex can be coupled using an in situ generated nickel catalyst to yield 3. Thus, a sterically hindered ortho-halopyrimidine site has undergone C-C coupling to give a bidentate diimine site. The true test of this synthetic approach was passed by binding a metal ion in the newly created bidentate site. The reaction of Ru(biquinoline)2Cl2 with 3 gave complex 4 as a green complex, confirming that different metal sub-units can be incorporated into the structure.
Although the creation of a new binding site is crucial to this approach, the steric constraints of the new binding site in 4 necessitated a more open ligand design. In an analogous approach, chloroterpyridine 5 was synthesized and complexed with the Ru(bpy)2 subunit in high yield to give 6. Subsequent coupling of 6 gave the binuclear complex 7, which contains a new binding site substantially removed from the other two sites.
Figure 5 Schematic representation of the second-generation polymetallic dendrimers. One of the crucial advantages of this approach is the economy of synthetic steps. The same chloro-substituted ligands are used in every growth phase of the dendrimer (Fig. 5a). By coupling the pentametallic complex in Figure 5b, a decanuclear complex would be obtained as shown in Figure 5c, allowing the cycle to continue. The methodology outlined above gives access to dendrimers with metal ions at the branching points of the dendrimer. The closed dendritic structures will contain 7 (Fig. 4f), 31 (Fig. 5f), and 127 (not shown) metal ions for the first, second and third generations, respectively. It is important to stress that the coupling reaction serves to create a new metal-binding site; a new binuclear "complex-ligand" (c.f., 4 and 7) is created starting from two mononuclear complexes. The penultimate branched materials containing 5 (Fig. 4e), 21 (Fig. 5e), and 85 (not shown) metal ions are of yet greater interest as these dendritic fragments are potent synthons that still contain two cis-metal coordination sites available for binding to different ligands. 2.1.2 Metal Ions at the Periphery of the DendrimerPlease contact me if you are interested in working with metallodendrimers. 2.2 The question of diastereomersOne major concern with polyruthenium dendrimers is that the complexes are synthesized as a mixture of isomers.8 This is due to the chirality associated with each octahedral metal surrounded by three bidentate ligands. Two possible solutions will be explored: 1) synthesizing new chiral ligands to form enantiopure starting materials; and 2) utilizing achiral tridentate building blocks. 2.2.1 Enantiopure complexesPlease contact me if you are interested in working with enantiopure metal complexes. 2.2.2 Achiral metal complexesTridentate ligands of the terpyridine type have previously been used in order to maintain achiral complexes.9 Therefore, a method of working with polymetallic complexes with a limited number of chiral centres has been developed. Mono- and bimetallic complexes containing tridentate terpyridine binding motifs shown on the right. The methodology to convert these molecules into polymetallic dendrimers will be the same as outlined in section 2.1.1. Upon complexation of the dimetallic complex around an octahedral metal ion, a heptametallic complex will be obtained (c.f., Fig. 4f). We have also introduced a tridentate site into the central position via Stille coupling methodology in order to maintain a fully achiral complex. 2.3 Room
temperature (r.t.) luminescence
|
|||||||
| Last Updated: December 20, 2004 | ||||||||
|
Copyright © 2004 Université de Montréal |
|

.gif)
.gif)